As an assessment for a unit in my Masters of Nutrition degree, I recently had to put together a ‘Factsheet’ on how our bodies make ATP (targeted at the level of a fellow student). I received a HD for this assessment piece and several of my friends expressed how interesting they found the factsheet. So I thought I’d share the information with you here!
For a bit of revision on Nutrients, try reading some of my previous blog posts:
Digestion and absorption of Lipids
ATP – Take a Tour of the Body’s Power Station
Energy for life
ENERGY enables us to be alive! Our bodies are like power stations, metabolising chemical energy from food into more usable, ‘high potential energy’ molecules of which Adenosine Triphosphate (ATP, Figure 1) is the most widely used [1]. ATP’s potential energy is found in the unstable bonds between its three phosphate groups[2]. ATP is used in the body in both catabolic (breaking down) and anabolic (building) reactions where these bonds break, releasing the potential energy, converting it to other types of energy[3].

Fig 1. ATP [2]
From blowing your nose (kinetic and and sound energy), maintaining body temperature (heat energy) or sending nerve impulses (electrical energy), ATP is being used. Disfunction in its synthesis can be caused by nutrient deficiencies, poisons, diseases even genetic mutations leading to many debilitating conditions, even death[4].
ATP (ENERGY) from Glucose
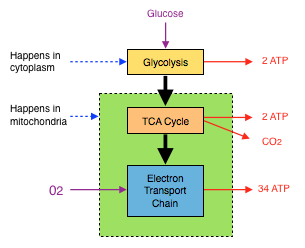
Every cell in the body is able to convert glucose into ATP[3]. Carbohydrates in food are digested and broken down into their monosaccharide components; glucose, fructose and galactose which enter the bloodstream. While fructose and galactose head to the liver for metabolism, glucose (with the help of insulin) is distributed to the body’s cells where it is either converted to glycogen for storage in muscles or liver, or used to produce ATP[3]. ATP is manufactured from glucose in 3 steps; glycolysis, the TCA Cycle and the electron transport chain (ETC).

Fig 2 ATP Production Summary
Glycolysis
Glycolysis (Fig 3) is the transformation of 6-carbon glucose to 3-carbon pyruvate in the cytoplasm of a cell[2]. It involves several intermediates, is anaerobic and has a net yield of 2ATP molecules per glucose molecule. The coenzyme nicotinamide adenine dinucleotide (NAD) is an electron transporter (electrons are very reactive so need to be carried by other molecules) essential in glycolysis as well as TCA cycle[4]. It picks up the hydrogen atoms and their electrons and transports them (as NADH) to the ETC for ATP production. Glycolysis releases 2 NADH molecules for each glucose molecule[2].

Fig 3. Glycolysis [3]
Krebs cycle (aka The Citric Acid Cycle, TCA cycle)
3-carbon pyruvate produced in glycolysis then enters the mitochondrion[2], where a carboxylate group is replaced by a molecule of co-enzyme A (CoA), to become acetyl CoA[2]. The carboxylate group is released as CO2, making this process irreversible[3]. Acetyl CoA then enters the TCA Cycle (Fig 4) whereby it joins with oxaloacetate moving through a series of transformations, releasing CO2, 2 ATP molecules and many electrons (in NADH and another coenzyme FADH2, flavin adenine dinucleotide)[4] in a cycle that finishes with oxaloacetate. If more acetyl CoA is available then the cycle will continue.

Fig 4. The TCA Cycle [5]
Electron Transport Chain (ETC)
The ETC is the real power generator. NADH and FADH2 from glycolysis and TCA cycle carry electrons to a series of enzymes called cytochromes on the inner mitochondrial membrane (see Fig 4.)[1]. Electrons flow down this cytochrome ‘chain’, as they would an electrical wire, while H+ is transferred across the membrane causing a concentration gradient[4]. Cytochrome C oxidase complex marks the end of the ETC transferring electrons to O2 producing water with H+ ions[1]. The H+ concentration gradient created is used to synthesise ATP through channel protein ATP Synthase. The movement of H+ through ATP synthase causes the protein to physically rotate, like a motor, forcing ADP (adenosine diphosphate) to bond with a phosphate ion, creating ATP[2]. Making 32-38 ATP molecules per glucose molecule, depending on the study[1,2,5].

Fig 5. Electron Transport Chain [1]
ATP from other macronutrients
Fructose and galactose are activated then enter the glycolysis pathway as intermediates (fructose-6-phosphate and glucose-6-phosphate respectively)[3]. Amino Acids are either glucogenic (can make glucose through gluconeogenesis) or ketogenic (catabolise to acetyl CoA and cannot be used to make glucose)[3]. After deamination, glycogenic amino acids form intermediates in glycolysis or the TCA cycle while ketogenic amino acids enter the pathway as acetyl CoA[2]. Fatty acids, like ketogenic amino acids, catabolise into acetyl CoA via beta-oxidation[2]. When fatty acids are in excess, acetyl CoA can exceed TCA cycle capacity. Excess acetyl CoA converts to ketones such as acetoacetate and β-hydroxy butyrate[6].

Fig 6 Macronutrient Energy Interrelationship, note arrow direction.
Can a Ketogenic Diet treat Epilepsy and Cancer?
Figure 5 illustrates the principle behind the use of a ketogenic diet as a mode of epilepsy treatment since the the early 1900s[6] and has recently to become a more widespread epilepsy treatment[7,8] and potential cancer treatment [9] .
Brain and nervous system cells are unable to store glucose (glycogen) therefore rely on blood glucose for energy metabolism[2]. In times of starvation ketone molecules can be converted into acetyl CoA in the mitochondria[2].
Ketogenic diets (high fat, adequate protein, low carbohydrate), mimic fasting hormonally, while meeting calorie intake[8]. Minimal carbohydrate results in little glycolysis, so energy must be made from ketones via acetyl CoA and the TCA cycle.
The precise mechanism by which this pathway soothes epileptic activity in neutrons is still not agreed upon. Some propose that glycolytic intermediates form precursors to epileptic reactions[6] so by eliminating glycolytic molecules in the cytoplasm, and shifting energy metabolism exclusively to the mitochondria, epileptic seizures are reduced.
The ketogenic diet’s success in treating some cancers also needs further mechanistic research. It is thought that certain cancer cells either lack mitochondria or are unable to use ketones as an energy source, relying solely on glucose. Thus, a ketogenic diet effectively starves cancer cells[9].
Nutrient Deficiencies
NAD and FAD are vital in ATP production, both are derived from essential dietary vitamins; niacin (B3) and riboflavin (B2) respectively. Thiamin (vitamin B1) forms the enzyme thiamin pyrophosphate (TPP), required in the decarboxylation of pyruvate to Acetyl CoA[3]. Deficiencies in any of these vitamins can lead to reduced ATP synthesis; nerve irritability/degeneration; beriberi (B1) where an excess in pyruvate (and lactate) cause pooling and oedema; and the 4 D’s of pellagra (B3 deficiency; dermatitis, diarrhoea, dementia and death)[2,3]. Deficiency in Australia is rare due to the presence of these vitamins in a wide range of nutritious foods.
References
1. Betts, G., Anatomy and Physiology. 2013, Houston, Texas, USA: Openstax College, Rice University.
2. Whitney, E.N., et al., Understanding nutrition. 2014: 2nd edition.
3. Crowe, T., HSN701 Study Guide. 2015, Deakin University: Burwood, Vic, Australia.
4. Parker, P., HSN749 Study Guide. 2016, Deakin University: Burwood, Vic, Australia.
5. VanPutte, C., et al., Seeley’s anatomy & physiology. 2013: McGraw-Hill Higher Education.
6. Noebels JL, A.M., Rogawski MA, Jasper’s Basic Mechanisms of the Epilepsies. 4th ed. 2012, Bethesda (MD): National Centre for Biotechnology Information (US).
7. Neal, E.G., et al., A randomized trial of classical and medium-chain triglyceride ketogenic diets in the treatment of childhood epilepsy. Epilepsia, 2009. 50(5): p. 1109-17.
8. Kossoff, E.H., et al., Diet Redux: Outcomes from Reattempting Dietary Therapy for Epilepsy. J Child Neurol, 2016.
9. Branco, A.F., et al., Ketogenic diets: from cancer to mitochondrial diseases and beyond. Eur J Clin Invest, 2016. 46(3): p. 285-98.